 |
| Previous Image | Next Image |
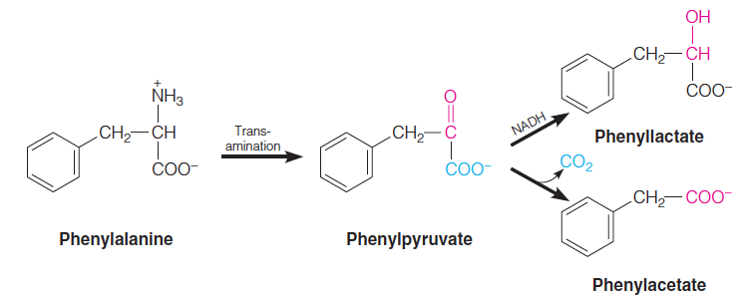
| Description: A hereditary deficiency of phenylalanine hydroxylase is responsible for phenylketonuria (PKU) In PKU, phenylalanine accumulates to very high levels (hyperphenylalaninem ia) because of the block in conversion to tyrosine, and much of this phenylalanine is metabolized via pathways that are normally little used—particularly transamination to phenylpyruvate (a phenylketone), and also subsequent conversion of phenylpyruvate to phenyllactate and phenylacetate. These compounds are excreted in urine in enormous quantities (1 to 2 grams per day). Picture Stats: Views: 164 Filesize: 52.37kB Height: 297 Width: 738 Source: https://biology-forums.com/index.php?action=gallery;sa=view;id=34775 |